Les dystrophies rétiniennes héréditaires
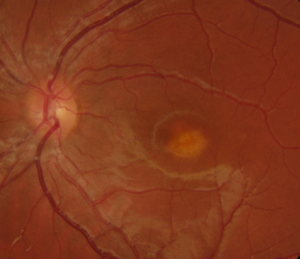 Les dystrophies rétiniennes héréditaires sont des maladies rares, de transmission familiale génétique pour la plupart et entrainant une perturbation du fonctionnement des cellules de la rétine (cônes et/ou bâtonnets) en atteignent préférentiellement la rétine centrale ou périphérique à l’origine de troubles visuels variables. Elles peuvent survenir à tout âge.
Les dystrophies rétiniennes héréditaires sont des maladies rares, de transmission familiale génétique pour la plupart et entrainant une perturbation du fonctionnement des cellules de la rétine (cônes et/ou bâtonnets) en atteignent préférentiellement la rétine centrale ou périphérique à l’origine de troubles visuels variables. Elles peuvent survenir à tout âge.
Les dystrophies les plus fréquentes sont :
- la rétinopathie pigmentaire,
- la dystrophie des cônes,
- la maladie de Stargardt,
- la maladie de Best.
Selon le type de dystrophie et le degré d’atteinte, les symptômes sont très variables selon les individus, d’une baisse de vision prédominant au niveau de la vision centrale (dystrophie des cônes, maladies de Best et de Stargardt) et/ou de la vision périphérique (rétinopathie pigmentaire), un trouble de la vision des couleurs, une nécessité de plus de lumière pour lire ou à une perturbation de la vision nocturne.
Le diagnostic repose sur : l’interrogatoire et l’arbre généalogique, l’examen du fond d’œil, le champ visuel central ou périphérique, les clichés en autofluorescence, l’angiographie à la fluorescéine et au vert d’infracyanine, les examens d’électrophysiologie: éléctrorétinogramme (ERG), electro-oculogramme (EOG) et potenteils évoqués visuels (PEV) et une éventuelle analyse génétique.
Il n’existe pas de traitement curatif à l’heure actuelle.